

Biotherapies applied to neuromuscular disabilities (LIA BAHN)
Composition de l'équipe
Directeur de Recherche CNRS
Ingénieur
Activities
{kind=link}
{kind=link}
Création du LIA BAHN
Le 14 novembre 2013, Mr. Jean-Luc Vayssière, Président de l'Université de Versailles Saint-Quentin-en-Yvelines (UVSQ), et Mr. Patrick Rampal, Président du Conseil d’Administration du Centre Scientifique de Monaco (CSM), ont signé une convention pour la création d’un Laboratoire International Associé dédié aux Biothérapies Appliquées aux Handicaps Neuromusculaires (LIA BAHN).
La convention LIA BAHN va permettre de réunir des scientifiques du CSM et de l'Équipe "Biothérapies des maladies du système neuromusculaire" dirigée par le Dr Luis Garcia de l’UVSQ au sein d'un Laboratoire International Associé (LIA).
Les objectifs de ce LIA établi au sein du CSM sont les suivants :
Ces recherches s’appuieront sur les plateformes technologiques de l’UFR des Sciences de la Santé Simone Veil de l’UVSQ et celles installées au sein du CSM, ainsi que sur de fortes collaborations internationales.
Ce LIA s’inscrit pleinement dans la dynamique de mise en place de collaboration de recherche – ici, dans le domaine des sciences du vivant - définie par l’UVSQ dans son contrat quinquennal, d’une part, et dans le cadre de l’émergence d’un nouvel axe de recherche en biologie médicale en principauté de Monaco selon les volontés de S.A.S. le Prince Albert II, d’autre part.
Par ailleurs, le soutien de longue date de l’Equipe par l’Association Monégasque contre les Myopathies, par le biais de son Président Mr. Luc Pettavino, a grandement contribué à l’établissement du LIA BAHN et à la mise en place d’une activité de recherche innovante sur les maladies neuromusculaires d’origine génétique en Principauté de Monaco.
Activités scientifiques du LIA BAHN
1. Stratégies thérapeutiques pour lutter contre la Dystrophie Musculaire de Duchenne (DMD)
Descriptif général de la pathologie.
Les maladies neuromusculaires regroupent un ensemble de plusieurs centaines de maladies, principalement d'origine génétique, définies par un défaut de commande du muscle ou par une destruction du tissu musculaire. Conjointement, elles affectent plusieurs dizaines de milliers de personnes en France et constituent un enjeu majeur de santé publique. La plus emblématique d'entre elles, la Dystrophie Musculaire de Duchenne (DMD) est causée par des mutations qui affectent le gène codant pour la dystrophine, une protéine indispensable au bon fonctionnement des cellules musculaires.

La dystrophine : du gène à la protéine.
Le gène codant pour la dystrophine est situé sur le bras court du chromosome X (locus 21.2). Au sein des muscles squelettiques, la protéine se localise sous la membrane des fibres musculaires et permet de créer un pont entre les filaments d’actine intracellulaires et les protéines extracellulaires de liaison à la lame basale (sarcoglycans, dystroglycans). Sur la partie de droite sont représentées deux coupes histologiques transversales d’un muscle squelettique. La microphotographie de gauche (coloration de coupes à l’hématéine-éosine) montre l’organisation des fibres musculaires, jointives et caractérisées par une forme parallélépipédique, dans lesquelles on peut observer certains noyaux de fibres (points violets) localisées en périphérie, ainsi que quelques vaisseaux sanguins (en haut à gauche). La microphotographie de droite représente une coupe similaire à la précédente marquée avec un anticorps spécifique de la dystrophine et révélé par émission de fluorescence verte. Cet immunomarquage confirme la localisation membranaire de la dystrophine. Les interactions entre la dystrophine et ses protéines associées confèrent aux fibres musculaires une plus grande résistance face aux nombreux évènements de contraction/relâchement caractéristiques de la physiologie du muscle squelettique. Chez les patients DMD, l’absence de dystrophine fragilise les fibres musculaires qui finissent par dégénérer progressivement au cours du temps.
La nature des mutations génétiques chez DMD.
Les maladies dites génétiques sont dues à des altérations de l’information génétique contenue le long de l’ADN, constituant fondamental des chromosomes. Elles peuvent provenir d’altérations de grande ampleur telle que des translocations chromosomiques, de l’apparition de chromosomes surnuméraires (cas des trisomies par exemple), ou de délétions de larges séquences d’ADN. Elles peuvent également être dues à des évènements plus fins tels que des pertes ou des substitutions d’une ou de quelques bases de l’ADN, pouvant provoquer diverses altérations moléculaires ou biochimiques. La conséquence la plus fréquente de ces altérations est le dysfonctionnement des protéines suivie de leur dégradation.
DMD n’est pas causée par un seul type de mutation génétique. Certaines formes sont liées à des mutations non-sens qui provoquent un arrêt prématuré de la synthèse de la protéine dystrophine, d’autres sont dues à des duplications de séquences ou encore à des mutations d’épissage qui conduisent à la transcription d’une information génétique erronée qui n’a plus rien à voir avec la séquence d’origine. Cependant, la grande majorité des mutations humaines de DMD sont causées par des délétions d’exons qui conduisent à un décalage du cadre de lecture de la séquence d’origine (voir schéma explicatif ci-dessous).

Représentation schématique de la notion de décalage du cadre de lecture au niveau de l’ADN génomique.
Dans cet exemple, nous considérons une séquence d’ADN normale à quatre exons (schéma du haut). Ces derniers sont représentés sous forme de « blocs » qui s’emboîtent les uns avec les autres afin de garantir la succession correcte des bases de l’ADN (A, T, C, G) et donc des acides aminés constituants la protéine issue de la transcription/traduction de l’information génétique. A chaque triplet de base correspond un acide aminé particulier conformément au code génétique universel. Notons cependant que la succession des bases (et donc des triplets) n’est pas toujours un multiple de trois au sein d’un exon donné. Par exemple à la frontière entre l’exon 1 et l’exon 2 la dernière base de l’exon 1 (A) nécessite les deux premières bases de l’exon 2 (GG) pour former le triplet suivant (AGG, codant pour Arg). La même remarque peut être faite à la jonction entre les exons 3 et 4. Dans le cas d’une délétion de l’exon 2 (schéma du milieu), les « blocs » exon1 et exon 3 ne sont plus en phase (les « blocs » ne s’emboîtent pas) puisque le premier triplet de l’exon 3 (TGG) qui devait coder pour un acide aminé à part entière (Trp) se retrouve « décalé » d’une base au niveau de la nouvelle frontière exon 1/exon 3 créée par la délétion. La dernière base de l’exon 1 (A) accapare en quelque sorte les deux premières bases de l’exon 3 (TG) pour former le triplet suivant (ATG) codant pour un acide aminé différent de l’original (Met à la place de Arg). Plus problématique est le décalage du cadre de lecture opéré par ce changement qui modifie totalement la séquence protéique en aval de l’exon 1 (acides aminés rouges sur le schéma du milieu). Cette modification de la séquence protéique, due au décalage du cadre de lecture original, génère une protéine non fonctionnelle, généralement rapidement dégradée. Comme nous le verrons par la suite pour DMD, le saut d’exon thérapeutique peut être utilisé pour rétablir un cadre de lecture correct en aval de la délétion. Dans notre exemple, nous observons que le saut induit de l’exon 3 (dans le cas d’un génome sans exon 2) permettrait théoriquement la réhabilitation d’un cadre de lecture normal entre l’exon 1 et l’exon 4 (schéma du bas). Pour certaines protéines particulières comme la dystrophine, le raccourcissement de taille opéré par le saut d’exon permet toutefois de conserver leur fonctionnalité.
Appliqué au gène de la dystrophine, le décalage du cadre de lecture conduit au phénotype DMD. Le schéma ci-dessous résume la problématique du décalage du cadre de lecture dans le contexte de la dystrophine.

Cadre de lecture de l’ARN messager de la dystrophine.
Le schéma de gauche montre l’organisation des 79 exons du gène de la dystrophine représentés par des blocs qui s’emboîtent parfaitement les uns à la suite des autres afin de conserver un cadre de lecture correct donnant lieu à la synthèse d’une protéine fonctionnelle. Le schéma en haut à droite représente le génotype d’un individu DMD dont il manque l’exon 52 (mutation delta52). Il en résulte que les exons 51 et 53 ne s’emboîtent pas parfaitement, conduisant à un décalage systématique du cadre de lecture à partir du début de l’exon 53, et donc à une séquence erronée de la dystrophine. Ce décalage du cadre de lecture conduit à la synthèse d’une protéine non fonctionnelle qui est rapidement dégradée. Le schéma en bas à droite représente le génotype d’un patient dont il manque les exons 51 et 52 (delta51-52). Paradoxalement, la délétion de ces deux exons confère à l’individu une physiopathologie beaucoup plus modérée que celle de l’individu delta52. Cette pathologie, bien que ciblant le même gène, porte le nom de Dystrophie Musculaire de Becker (BMD en anglais). La raison conduisant à cette forme modérée de la maladie est due au fait que les exons 50 et 53 qui bornent le site muté s’emboîtent convenablement, permettant ainsi de conserver un cadre de lecture correct sur toute la séquence de l’ARNm à partir de l’exon 53. La protéine résultante, bien qu’écourtée, demeure fonctionnelle.
L’approche de saut d’exon thérapeutique.
L’observation d’une forme pathologique modérée chez les patients atteints de Dystrophie Musculaire de Becker (BMD) a conduit les chercheurs à envisager une voie thérapeutique originale : rétablir un cadre de lecture correct en parvenant à éliminer un ou plusieurs exons. Dans une première approche, cette action peut avoir lieu lors de la phase de maturation du pre-ARN vers l’ARN messager dystrophine. C’est en effet lors de cette étape de la transcription que les parties non codantes du pre-ARN, les introns, sont éliminées au cours du processus d’épissage. Le saut d’exon thérapeutique est donc une approche qui consiste à utiliser des outils moléculaires capables d’interférer avec la machinerie d’épissage de la cellule afin d’induire un épissage plus large capable d’éliminer un ou plusieurs exons ciblés afin de rétablir un cadre de lecture correct.

Mécanisme d’épissage et saut d’exon.
La partie gauche schématise le passage de l’information génétique contenue dans la molécule d’ADN à la synthèse d’une protéine. Le mécanisme nécessite une série de processus moléculaires qui débutent par la transcription de la séquence d’un gène contenue dans l’ADN en une copie appelée pre-ARN, qui subit elle-même une maturation (élimination des introns, parties non codantes du gène) pour former l’ARN messager (ARNm) : ce dernier mécanisme est appelé épissage. L’ARNm, produit dans le noyau cellulaire, est alors transporté dans le cytoplasme pour être traduit en protéine. La partie droite schématise le principe général du saut d’exon. Une séquence oligonucléotidique antisens est acheminée jusqu’au noyau cellulaire et se lie par complémentarité de bases sur une séquence de reconnaissance de la machinerie d’épissage (en rouge sur le schéma). L’encombrement stérique provoqué par la présence de la séquence antisens empêche le bon déroulement du processus d’épissage autour de ce site (dans cet exemple autour de l’exon 2). La machinerie d’épissage est en quelque sorte « contrainte » de se déplacer vers les séquences d’épissage situées en aval de celles qui sont masquées, et finit par provoquer un épissage plus large que d’habitude, englobant au passage le ou les exons situés à l’intérieur de la zone concernée (dans cet exemple, cela provoque le saut de l’exon 2).
Notons cependant que l’applicabilité du saut d’exon pour DMD est rendue possible par le fait que la dystrophine possède des domaines répétés de type spectrine (de couleur verte sur les représentations schématiques de la dystrophine) dont plusieurs peuvent être supprimés sans que cela n’affecte la fonctionnalité de la protéine (comme observé sur chez certains génotypes BMD). Cette stratégie est plus difficilement applicable sur d’autres protéines pour lesquelles il existe un risque élevé de perte de conformation et donc de fonctionnalité.
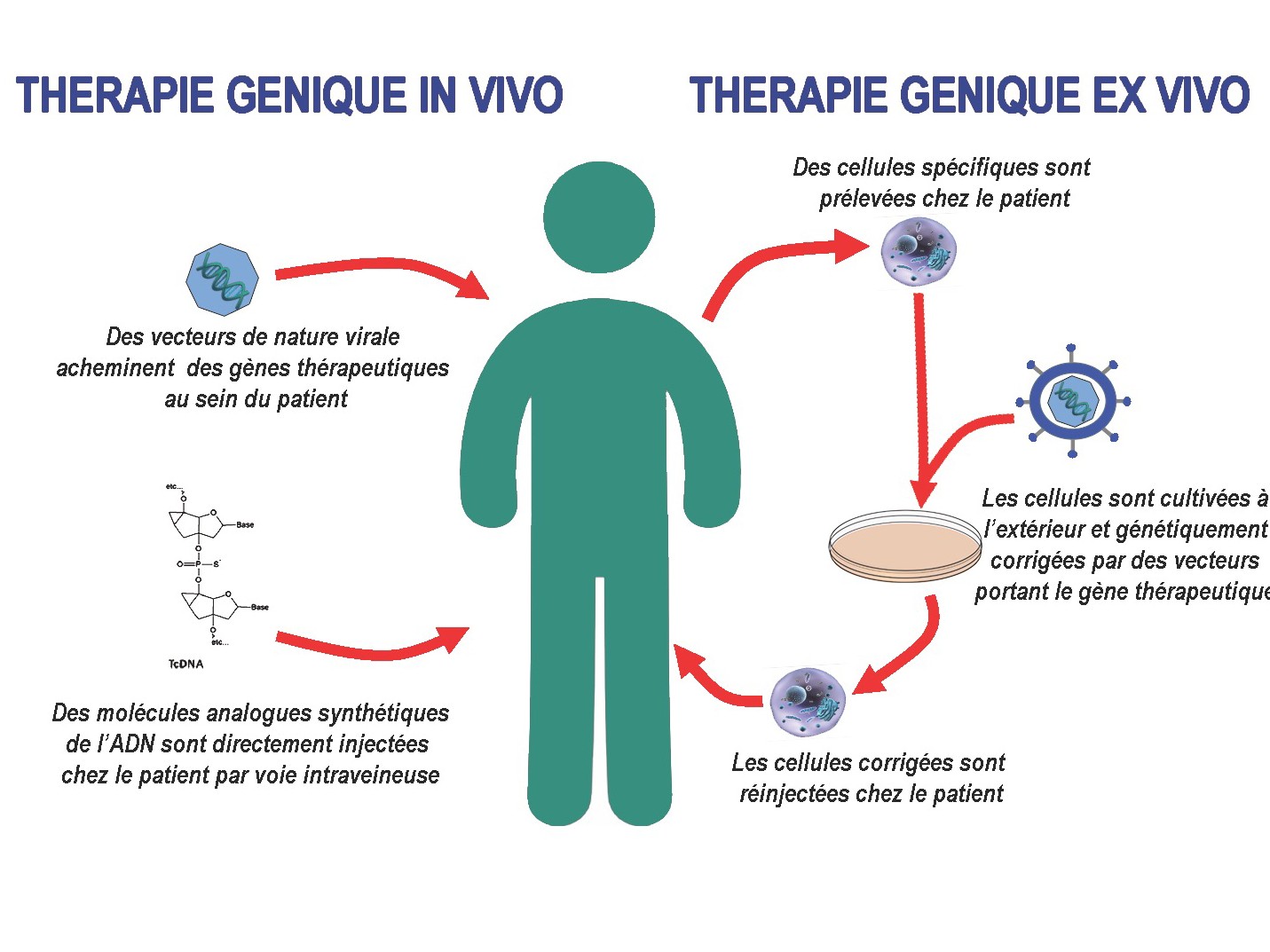
Les outils moléculaires que nous développons dans le cadre du saut d’exon sont de deux types : des petits ARN nucléaires dérivés de l’U7snRNA (small nuclear RNA) et des molécules oligonucléotidiques antisens chimiquement modifiés appelées tricyclo-DNA (tcDNA).
• L’outil U7snRNA
Les U7snRNA sont des petits ARN nucléaires impliqués dans la maturation des pré-ARN histones. Ils forment un complexe ribnucléoprotéique appelé U7snRNP (small nuclear RiboNucleoProtein). Les U7snRNA possèdent une séquence ARN complémentaire d’une partie 3’ des pré-ARN histone et en s’y fixant, ils favorisent l’accroche d’un complexe protéique qui finit par cliver la partie 3’ des pré-ARN histone.

Structure des U7snRNA et mode de liaison sur le pré-ARN dystrophine.
Le schéma de gauche représente la structure d’un U7snRNP sur lequel la partie grisée en 5’ correspond à la partie complémentaire des pré-ARN histone (séquence HDE : Histone Downstream Element). La partie 3’ prend une conformation en 3’ indispensable pour garantir la fonctionnalité de la molécule. Le complexe protéique lié à l’U7snRNA est représenté sous forme d’un anneau constitué de 7 protéines différentes. Le schéma de droite représente l’U7snRNP dont la séquence HDE a été modifiée pour cibler un ou plusieurs sites de reconnaissance de la machinerie d’épissage. En s’y fixant, l’U7snRNP induit le saut d’exon souhaité.
Une étude publiée en 2004 par l’équipe dans la prestigieuse revue Science avait apportée la preuve de principe expérimentale de l’efficacité de l’U7snRNA modifié à corriger le phénotype dystrophique dans le modèle de souris mdx (voir Goyenvalle et al, Science, 2004).
• L’outil tricyclo-DNA
Il est connu de longue date que le transfert cellulaire de molécules d’oligonucléotides ou d’oligoribonucléotides n’est pas efficace car ces molécules sans coiffe 5’ ni signal de polyadénylation sont rapidement dégradées par des nucléases intracellulaires. Depuis quelques années un champs d’investigation important s’est développé à l’interface chimie/biologie par la synthèse de molécules oligonucléotidiques chimiquement modifiées pour échapper ou résister à l’attaque des nucléases.

Représentation des diverses chimies d’oligonucléotides utilisées dans les stratégies de saut d’exon.
La structure de la molécule d’ADN classique est représentée dans la partie supérieure gauche. Les autres chimies existantes sont représentées sur la partie droite (RNase H-independent). Notre molécule exclusive, le tricyclo-DNA (Tc-DNA) est montrée en bas à droite.
Une étude récente publiée dans Nature Medecine a apporté la preuve de principe que la chimie Tc-DNA possédait des avantages certains comparativement aux autres chimies, notamment une capacité plus grande à franchir la barrière hémato-encéphalique donnant accès directe au cerveau (voir Goyenvalle et al, Nature Medecine, 2016). Les études actuelles portent sur les études de toxicité pour définir les doses et les fréquences d’injections les plus optimales d’un point de vu thérapeutique et engendrant un minimum d’effets secondaires.
L’approche d’édition génomique.
Une autre approche thérapeutique est actuellement testée par l’équipe : l’édition génomique par utilisation de l’outil CRISPR-Cas9. L’édition génomique regroupe les stratégies visant à induire des modifications génétiques non plus au niveau ARN mais directement à la source, c’est-à-dire au niveau de l’ADN génomique. Pour DMD, deux voies intéressantes s’offrent aux chercheurs : un saut d’exon induit sur l’ADN même ou une réparation de la mutation par le mécanisme de la recombinaison homologue.
2. Projet interface : production de protéines recombinantes issues du milieu marin
Notre savoir-faire technologique dans la production de vecteurs viraux de thérapie génique à grande échelle nous a conduits à initier des collaborations avec les équipes de biologie marine afin de répondre à leur besoin d’étudier plus en détail la structure et par delà, la fonction de protéines issues du monde marin, et notamment du corail.
D’un point de vue méthodologique, nous produisons les protéines recombinantes par le système baculovirus/cellules d’insecte.

Représentation schématique du procédé de production de protéines recombinantes ou de vecteurs viraux par le système baculovirus/cellules d’insectes.
L’étape 1 consiste à produire des vecteurs baculoviraux recombinants pour la protéine d’intérêt. Les étapes suivantes consistent à amplifier la production baculovirale par des passages successifs dans des cellules d’insecte. Les baculovirus étant infectieux et réplicatifs, une réaction en chaîne a lieu au sein de la culture d’insecte, amplifiant par la même occasion la quantité de protéines recombinantes.
L’utilisation de plusieurs méthodes de clarification, concentration et purification des protéines nous permettent d’atteindre des niveaux de puretés suffisants pour pouvoir étudier plus en détail leur structure et/ou leur fonction.
Le 14 novembre 2013, Mr. Jean-Luc Vayssière, Président de l'Université de Versailles Saint-Quentin-en-Yvelines (UVSQ), et Mr. Patrick Rampal, Président du Conseil d’Administration du Centre Scientifique de Monaco (CSM), ont signé une convention pour la création d’un Laboratoire International Associé dédié aux Biothérapies Appliquées aux Handicaps Neuromusculaires (LIA BAHN).
 |
Signature de la convention LIA-BAHN. L’ex-Président de l’Université de Versailles Saint-Quentin en Yvelines, Mr. Jean-Luc Vayssière (assis à gauche) et le Président du Conseil d’Administration du Centre Scientifique de Monaco, Mr. Patrick Rampal (assis à droite), sous l’autorité du Président de la République Française, Mr. François Hollande et de S.A.S. le Prince Albert II de Monaco, le jour de la signature de la convention LIA-BAHN, le 14 novembre 2013. |
Les objectifs de ce LIA établi au sein du CSM sont les suivants :
- La consolidation et le développement d’une recherche translationnelle innovante pour des maladies dont les besoins thérapeutiques ne sont pas encore couverts.
- La création de synergies et la coopération pluridisciplinaire par le co-développement de nouveaux outils moléculaires issus des biotechnologies pour le traitement de maladies génétiques et/ou oncologiques.
- La mise en place de collaborations internes visant à développer des projets « interfaces » originaux entre les équipes de biologie médicale et les équipes de biologie marine.
Ces recherches s’appuieront sur les plateformes technologiques de l’UFR des Sciences de la Santé Simone Veil de l’UVSQ et celles installées au sein du CSM, ainsi que sur de fortes collaborations internationales.
Ce LIA s’inscrit pleinement dans la dynamique de mise en place de collaboration de recherche – ici, dans le domaine des sciences du vivant - définie par l’UVSQ dans son contrat quinquennal, d’une part, et dans le cadre de l’émergence d’un nouvel axe de recherche en biologie médicale en principauté de Monaco selon les volontés de S.A.S. le Prince Albert II, d’autre part.
Par ailleurs, le soutien de longue date de l’Equipe par l’Association Monégasque contre les Myopathies, par le biais de son Président Mr. Luc Pettavino, a grandement contribué à l’établissement du LIA BAHN et à la mise en place d’une activité de recherche innovante sur les maladies neuromusculaires d’origine génétique en Principauté de Monaco.
 |
Equipe du LIA BAHN lors de l’inauguration officielle des locaux par S.A.S. le Prince Albert II de Monaco le 01 octobre 2014. De gauche à droite : Christian Delporte (UVSQ), Luis Garcia (Directeur LIA BAHN), Valérie Robin, Luc Pettavino (Président de l’Association Monégasque contre les Myopathies), Aurélie Avril, Pierre-Olivier Buclez, Monique Pettavino, Cyriaque Beley, S.A.S. le Prince Albert II de Monaco, Rachid Benchaouir, Graziella Griffith et Aurélie Goyenvalle. |
1. Stratégies thérapeutiques pour lutter contre la Dystrophie Musculaire de Duchenne (DMD)
Descriptif général de la pathologie.
Les maladies neuromusculaires regroupent un ensemble de plusieurs centaines de maladies, principalement d'origine génétique, définies par un défaut de commande du muscle ou par une destruction du tissu musculaire. Conjointement, elles affectent plusieurs dizaines de milliers de personnes en France et constituent un enjeu majeur de santé publique. La plus emblématique d'entre elles, la Dystrophie Musculaire de Duchenne (DMD) est causée par des mutations qui affectent le gène codant pour la dystrophine, une protéine indispensable au bon fonctionnement des cellules musculaires.
La dystrophine : du gène à la protéine.
Le gène codant pour la dystrophine est situé sur le bras court du chromosome X (locus 21.2). Au sein des muscles squelettiques, la protéine se localise sous la membrane des fibres musculaires et permet de créer un pont entre les filaments d’actine intracellulaires et les protéines extracellulaires de liaison à la lame basale (sarcoglycans, dystroglycans). Sur la partie de droite sont représentées deux coupes histologiques transversales d’un muscle squelettique. La microphotographie de gauche (coloration de coupes à l’hématéine-éosine) montre l’organisation des fibres musculaires, jointives et caractérisées par une forme parallélépipédique, dans lesquelles on peut observer certains noyaux de fibres (points violets) localisées en périphérie, ainsi que quelques vaisseaux sanguins (en haut à gauche). La microphotographie de droite représente une coupe similaire à la précédente marquée avec un anticorps spécifique de la dystrophine et révélé par émission de fluorescence verte. Cet immunomarquage confirme la localisation membranaire de la dystrophine. Les interactions entre la dystrophine et ses protéines associées confèrent aux fibres musculaires une plus grande résistance face aux nombreux évènements de contraction/relâchement caractéristiques de la physiologie du muscle squelettique. Chez les patients DMD, l’absence de dystrophine fragilise les fibres musculaires qui finissent par dégénérer progressivement au cours du temps.
La nature des mutations génétiques chez DMD.
Les maladies dites génétiques sont dues à des altérations de l’information génétique contenue le long de l’ADN, constituant fondamental des chromosomes. Elles peuvent provenir d’altérations de grande ampleur telle que des translocations chromosomiques, de l’apparition de chromosomes surnuméraires (cas des trisomies par exemple), ou de délétions de larges séquences d’ADN. Elles peuvent également être dues à des évènements plus fins tels que des pertes ou des substitutions d’une ou de quelques bases de l’ADN, pouvant provoquer diverses altérations moléculaires ou biochimiques. La conséquence la plus fréquente de ces altérations est le dysfonctionnement des protéines suivie de leur dégradation.
DMD n’est pas causée par un seul type de mutation génétique. Certaines formes sont liées à des mutations non-sens qui provoquent un arrêt prématuré de la synthèse de la protéine dystrophine, d’autres sont dues à des duplications de séquences ou encore à des mutations d’épissage qui conduisent à la transcription d’une information génétique erronée qui n’a plus rien à voir avec la séquence d’origine. Cependant, la grande majorité des mutations humaines de DMD sont causées par des délétions d’exons qui conduisent à un décalage du cadre de lecture de la séquence d’origine (voir schéma explicatif ci-dessous).
Représentation schématique de la notion de décalage du cadre de lecture au niveau de l’ADN génomique.
Dans cet exemple, nous considérons une séquence d’ADN normale à quatre exons (schéma du haut). Ces derniers sont représentés sous forme de « blocs » qui s’emboîtent les uns avec les autres afin de garantir la succession correcte des bases de l’ADN (A, T, C, G) et donc des acides aminés constituants la protéine issue de la transcription/traduction de l’information génétique. A chaque triplet de base correspond un acide aminé particulier conformément au code génétique universel. Notons cependant que la succession des bases (et donc des triplets) n’est pas toujours un multiple de trois au sein d’un exon donné. Par exemple à la frontière entre l’exon 1 et l’exon 2 la dernière base de l’exon 1 (A) nécessite les deux premières bases de l’exon 2 (GG) pour former le triplet suivant (AGG, codant pour Arg). La même remarque peut être faite à la jonction entre les exons 3 et 4. Dans le cas d’une délétion de l’exon 2 (schéma du milieu), les « blocs » exon1 et exon 3 ne sont plus en phase (les « blocs » ne s’emboîtent pas) puisque le premier triplet de l’exon 3 (TGG) qui devait coder pour un acide aminé à part entière (Trp) se retrouve « décalé » d’une base au niveau de la nouvelle frontière exon 1/exon 3 créée par la délétion. La dernière base de l’exon 1 (A) accapare en quelque sorte les deux premières bases de l’exon 3 (TG) pour former le triplet suivant (ATG) codant pour un acide aminé différent de l’original (Met à la place de Arg). Plus problématique est le décalage du cadre de lecture opéré par ce changement qui modifie totalement la séquence protéique en aval de l’exon 1 (acides aminés rouges sur le schéma du milieu). Cette modification de la séquence protéique, due au décalage du cadre de lecture original, génère une protéine non fonctionnelle, généralement rapidement dégradée. Comme nous le verrons par la suite pour DMD, le saut d’exon thérapeutique peut être utilisé pour rétablir un cadre de lecture correct en aval de la délétion. Dans notre exemple, nous observons que le saut induit de l’exon 3 (dans le cas d’un génome sans exon 2) permettrait théoriquement la réhabilitation d’un cadre de lecture normal entre l’exon 1 et l’exon 4 (schéma du bas). Pour certaines protéines particulières comme la dystrophine, le raccourcissement de taille opéré par le saut d’exon permet toutefois de conserver leur fonctionnalité.
Appliqué au gène de la dystrophine, le décalage du cadre de lecture conduit au phénotype DMD. Le schéma ci-dessous résume la problématique du décalage du cadre de lecture dans le contexte de la dystrophine.
Cadre de lecture de l’ARN messager de la dystrophine.
Le schéma de gauche montre l’organisation des 79 exons du gène de la dystrophine représentés par des blocs qui s’emboîtent parfaitement les uns à la suite des autres afin de conserver un cadre de lecture correct donnant lieu à la synthèse d’une protéine fonctionnelle. Le schéma en haut à droite représente le génotype d’un individu DMD dont il manque l’exon 52 (mutation delta52). Il en résulte que les exons 51 et 53 ne s’emboîtent pas parfaitement, conduisant à un décalage systématique du cadre de lecture à partir du début de l’exon 53, et donc à une séquence erronée de la dystrophine. Ce décalage du cadre de lecture conduit à la synthèse d’une protéine non fonctionnelle qui est rapidement dégradée. Le schéma en bas à droite représente le génotype d’un patient dont il manque les exons 51 et 52 (delta51-52). Paradoxalement, la délétion de ces deux exons confère à l’individu une physiopathologie beaucoup plus modérée que celle de l’individu delta52. Cette pathologie, bien que ciblant le même gène, porte le nom de Dystrophie Musculaire de Becker (BMD en anglais). La raison conduisant à cette forme modérée de la maladie est due au fait que les exons 50 et 53 qui bornent le site muté s’emboîtent convenablement, permettant ainsi de conserver un cadre de lecture correct sur toute la séquence de l’ARNm à partir de l’exon 53. La protéine résultante, bien qu’écourtée, demeure fonctionnelle.
L’approche de saut d’exon thérapeutique.
L’observation d’une forme pathologique modérée chez les patients atteints de Dystrophie Musculaire de Becker (BMD) a conduit les chercheurs à envisager une voie thérapeutique originale : rétablir un cadre de lecture correct en parvenant à éliminer un ou plusieurs exons. Dans une première approche, cette action peut avoir lieu lors de la phase de maturation du pre-ARN vers l’ARN messager dystrophine. C’est en effet lors de cette étape de la transcription que les parties non codantes du pre-ARN, les introns, sont éliminées au cours du processus d’épissage. Le saut d’exon thérapeutique est donc une approche qui consiste à utiliser des outils moléculaires capables d’interférer avec la machinerie d’épissage de la cellule afin d’induire un épissage plus large capable d’éliminer un ou plusieurs exons ciblés afin de rétablir un cadre de lecture correct.
Mécanisme d’épissage et saut d’exon.
La partie gauche schématise le passage de l’information génétique contenue dans la molécule d’ADN à la synthèse d’une protéine. Le mécanisme nécessite une série de processus moléculaires qui débutent par la transcription de la séquence d’un gène contenue dans l’ADN en une copie appelée pre-ARN, qui subit elle-même une maturation (élimination des introns, parties non codantes du gène) pour former l’ARN messager (ARNm) : ce dernier mécanisme est appelé épissage. L’ARNm, produit dans le noyau cellulaire, est alors transporté dans le cytoplasme pour être traduit en protéine. La partie droite schématise le principe général du saut d’exon. Une séquence oligonucléotidique antisens est acheminée jusqu’au noyau cellulaire et se lie par complémentarité de bases sur une séquence de reconnaissance de la machinerie d’épissage (en rouge sur le schéma). L’encombrement stérique provoqué par la présence de la séquence antisens empêche le bon déroulement du processus d’épissage autour de ce site (dans cet exemple autour de l’exon 2). La machinerie d’épissage est en quelque sorte « contrainte » de se déplacer vers les séquences d’épissage situées en aval de celles qui sont masquées, et finit par provoquer un épissage plus large que d’habitude, englobant au passage le ou les exons situés à l’intérieur de la zone concernée (dans cet exemple, cela provoque le saut de l’exon 2).
Notons cependant que l’applicabilité du saut d’exon pour DMD est rendue possible par le fait que la dystrophine possède des domaines répétés de type spectrine (de couleur verte sur les représentations schématiques de la dystrophine) dont plusieurs peuvent être supprimés sans que cela n’affecte la fonctionnalité de la protéine (comme observé sur chez certains génotypes BMD). Cette stratégie est plus difficilement applicable sur d’autres protéines pour lesquelles il existe un risque élevé de perte de conformation et donc de fonctionnalité.
Les outils moléculaires que nous développons dans le cadre du saut d’exon sont de deux types : des petits ARN nucléaires dérivés de l’U7snRNA (small nuclear RNA) et des molécules oligonucléotidiques antisens chimiquement modifiés appelées tricyclo-DNA (tcDNA).
• L’outil U7snRNA
Les U7snRNA sont des petits ARN nucléaires impliqués dans la maturation des pré-ARN histones. Ils forment un complexe ribnucléoprotéique appelé U7snRNP (small nuclear RiboNucleoProtein). Les U7snRNA possèdent une séquence ARN complémentaire d’une partie 3’ des pré-ARN histone et en s’y fixant, ils favorisent l’accroche d’un complexe protéique qui finit par cliver la partie 3’ des pré-ARN histone.
Structure des U7snRNA et mode de liaison sur le pré-ARN dystrophine.
Le schéma de gauche représente la structure d’un U7snRNP sur lequel la partie grisée en 5’ correspond à la partie complémentaire des pré-ARN histone (séquence HDE : Histone Downstream Element). La partie 3’ prend une conformation en 3’ indispensable pour garantir la fonctionnalité de la molécule. Le complexe protéique lié à l’U7snRNA est représenté sous forme d’un anneau constitué de 7 protéines différentes. Le schéma de droite représente l’U7snRNP dont la séquence HDE a été modifiée pour cibler un ou plusieurs sites de reconnaissance de la machinerie d’épissage. En s’y fixant, l’U7snRNP induit le saut d’exon souhaité.
Une étude publiée en 2004 par l’équipe dans la prestigieuse revue Science avait apportée la preuve de principe expérimentale de l’efficacité de l’U7snRNA modifié à corriger le phénotype dystrophique dans le modèle de souris mdx (voir Goyenvalle et al, Science, 2004).
• L’outil tricyclo-DNA
Il est connu de longue date que le transfert cellulaire de molécules d’oligonucléotides ou d’oligoribonucléotides n’est pas efficace car ces molécules sans coiffe 5’ ni signal de polyadénylation sont rapidement dégradées par des nucléases intracellulaires. Depuis quelques années un champs d’investigation important s’est développé à l’interface chimie/biologie par la synthèse de molécules oligonucléotidiques chimiquement modifiées pour échapper ou résister à l’attaque des nucléases.
Représentation des diverses chimies d’oligonucléotides utilisées dans les stratégies de saut d’exon.
La structure de la molécule d’ADN classique est représentée dans la partie supérieure gauche. Les autres chimies existantes sont représentées sur la partie droite (RNase H-independent). Notre molécule exclusive, le tricyclo-DNA (Tc-DNA) est montrée en bas à droite.
Une étude récente publiée dans Nature Medecine a apporté la preuve de principe que la chimie Tc-DNA possédait des avantages certains comparativement aux autres chimies, notamment une capacité plus grande à franchir la barrière hémato-encéphalique donnant accès directe au cerveau (voir Goyenvalle et al, Nature Medecine, 2016). Les études actuelles portent sur les études de toxicité pour définir les doses et les fréquences d’injections les plus optimales d’un point de vu thérapeutique et engendrant un minimum d’effets secondaires.
L’approche d’édition génomique.
Une autre approche thérapeutique est actuellement testée par l’équipe : l’édition génomique par utilisation de l’outil CRISPR-Cas9. L’édition génomique regroupe les stratégies visant à induire des modifications génétiques non plus au niveau ARN mais directement à la source, c’est-à-dire au niveau de l’ADN génomique. Pour DMD, deux voies intéressantes s’offrent aux chercheurs : un saut d’exon induit sur l’ADN même ou une réparation de la mutation par le mécanisme de la recombinaison homologue.
2. Projet interface : production de protéines recombinantes issues du milieu marin
Notre savoir-faire technologique dans la production de vecteurs viraux de thérapie génique à grande échelle nous a conduits à initier des collaborations avec les équipes de biologie marine afin de répondre à leur besoin d’étudier plus en détail la structure et par delà, la fonction de protéines issues du monde marin, et notamment du corail.
D’un point de vue méthodologique, nous produisons les protéines recombinantes par le système baculovirus/cellules d’insecte.
Représentation schématique du procédé de production de protéines recombinantes ou de vecteurs viraux par le système baculovirus/cellules d’insectes.
L’étape 1 consiste à produire des vecteurs baculoviraux recombinants pour la protéine d’intérêt. Les étapes suivantes consistent à amplifier la production baculovirale par des passages successifs dans des cellules d’insecte. Les baculovirus étant infectieux et réplicatifs, une réaction en chaîne a lieu au sein de la culture d’insecte, amplifiant par la même occasion la quantité de protéines recombinantes.
L’utilisation de plusieurs méthodes de clarification, concentration et purification des protéines nous permettent d’atteindre des niveaux de puretés suffisants pour pouvoir étudier plus en détail leur structure et/ou leur fonction.
Publications
2024
-
Identification of miR-141 as a Regulator of Epidermal Homeostasis
Vieu D L, Golebiewski C, Gastaldi C, Foucher A, Mari B, Rezzonico R, Droit A, Dumont M, Bastien P, Bernerd F, Marionnet C
J Invest Dermatol 145(7):1670-1682 -
The bench to bedside journey of tricyclo-DNA antisense oligonucleotides for the treatment of Duchenne muscular dystrophy
Blitek M, Phongsavanh X, Goyenvalle A
RSC Med Chem 15(9):3017-3025
2023
-
A group of novel VEGF splice variants as alternative therapeutic targets in renal cell carcinoma
Montemagno C, Durivault J, Gastaldi C, Dufies M, Vial V, He X, Ambrosetti D, Kamenskaya A, Négrier S, Bernhard J-C, Borchiellini D, Cao Y, Pagès G
mol 21 Feb 2023, 45 -
Dystrophin myonuclear domain restoration governs treatment efficacy in dystrophic muscle
Morin A, Stantzou A, Petrova O N, Hildyard J, Tensorer T, Matouk M, Petkova M V, Richard I, Manoliu T, Goyenvalle A, Falcone S, Schuelke M, Laplace-Builhé C, Piercy R J, Garcia L, Amthor H
Proc Natl Acad Sci USA 120(2):e2206324120 -
Identification and functional validation of SRC and RAPGEF1 as new direct targets of miR‐203, involved in regulation of epidermal homeostasis
Golebiewski C, Gastaldi C, Vieu D-L, Mari B, Rezzonico R, Bernerd F, Marionnet C
Sci Rep 13(1):14006 -
Oligonucleotide Enhancing Compound Increases Tricyclo-DNA Mediated Exon-Skipping Efficacy in the Mdx Mouse Model
Bizot F, Fayssoil A, Gastaldi C, Irawan T, Phongsavanh X, Mansart A, Tensorer T, Brisebard E, Garcia L, Juliano R L, Goyenvalle A
Cells 12(5):702
2022
-
Palmitic acid conjugation enhances potency of tricyclo-DNA splice switching oligonucleotides
Relizani K, Echevarria L, Zarrouki F, Gastaldi C, Dambrune C, Aupy P, Haeberli A, Komisarski M, Tensorer T, Larcher T, Svinartchouk F, Vaillend C, Garcia L, Goyenvalle A
nar Vol. 50, No. 1, 17–34 -
Partial Restoration of Brain Dystrophin and Behavioral Deficits by Exon Skipping in the Muscular Dystrophy X-Linked (mdx) Mouse
Zarrouki F, Relizani K, Bizot F, Tensorer T, Garcia L, Vaillend C, Goyenvalle A
Ana 92(2):213-229
2021
-
Altered visual processing in the mdx52 mouse model of Duchenne muscular dystrophy
Barboni M T S , Liber A M P, Joachimsthaler A, Saoudi A, Goyenvalle A, Rendon A, Roger J E , Ventura D F, Kremers J, Vaillend C
Neurobiol Dis 152: 105288 -
Association between prophylactic angiotensin-converting enzyme inhibitors and overall survival in Duchenne muscular dystrophy-analysis of registry data
Porcher R, Desguerre I, Amthor H, Chabrol B, Audic F, Rivier F, Isapof A, Tiffreau V, Campana-Salort E, Leturcq F, Tuffery-Giraud S, Ben Yaou R, Annane D, Amédro P, Barnerias C, Bécane H M, Béhin A, Bonnet D, Bassez G, Cossée M, de la Villéon G, Delcourte C, Fayssoil A, Fontaine B, Godart F, Guillaumont S, Jaillette E, Laforêt P, Leonard-Louis S, Lofaso F, Mayer M, Morales R J, Meune C, Orlikowski D, Ovaert C, Prigent H, Saadi M, Sochala M, Tard C, Vaksmann G, Walther-Louvier U, Eymard B, Stojkovic T, Ravaud P, Duboc D, Wahbi K
Eur Heart J 42(20):1976-1984 -
Les approches thérapeutiques de modulation de l'épissage - Avancées et perspectives
Saoudi A, Goyenvalle A
Med Sci (Paris) Vol. 37, N° 6-7: 625-631 -
RNA splicing modulation: Therapeutic progress and perspectives.
Saoudi A, Goyenvalle A
Med Sci 37(6-7):625-631
2020
-
Current Status of Antisense Oligonucleotide‑Based Therapy in Neuromuscular Disorders
Bizot F, Vulin A, Goyenvalle A
Drugs doi.org/10.1007/s40265-020-01363-3 -
Identifying and Avoiding tcDNA-ASO Sequence-Specific Toxicity for the Development of DMD Exon 51 Skipping Therapy
Aupy P, Echevarria L, Relizani K, Zarrouki F, Haeberli A, Komisarski M, Tensorer T, Jouvion G, Svinartchouk F, Garcia L, Goyenvalle A
Mol Ther Nucleic Acids 19: 371-383 -
Inhibition of Activin/Myostatin signalling induces skeletal muscle hypertrophy but impairs mouse testicular development
Vaughan D, Ritvos O, Mitchell R, Kretz O, Lalowski M, Amthor H, Chambers D, Matsakas A, Pasternack A, Collins-Hooper H, Ballesteros R, Huber T B, Denecke B, Widera D, Mukherjee A, Patel K
Eur J Transl Muol 30(1): 1-17 -
Live-imaging of revertant and therapeutically restored dystrophin in the DmdEGFP-mdx mouse model for Duchenne muscular dystrophy
Petkova M V, Stantzou A, Morin A, Petrova O, Morales-Gonzalez S, Seifert F, Bellec-Dyevre J, Manoliu T, Goyenvalle A, Garcia L, Richard I, Laplace-Builhé C, Schuelke M, Amthor H
Neuropathology and Applied Neurobiology doi: 10.1111/nan.12639 -
Long-Term Efficacy of AAV9-U7snRNA-Mediated Exon 51 Skipping in mdx52 Mice
Aupy P, Zarrouki F, Sandro Q, Gastaldi C, Buclez P-O, Mamchaoui K, Garcia L, Vaillend C, Goyenvalle A
Molecular Therapy: Methods & Clinical Development 17: 1037-1047 -
SIX1 and SIX4 homeoproteins regulate PAX7+ progenitor cell properties during fetal epaxial myogenesis
Wurmser M, Chaverot N, Madani R, Sakai H, Negroni E, Demignon J, Saint-Pierre B, Mouly V, Amthor H, Tapscott S, Birchmeier C, Tajbakhsh S, Le Grand F, Sotiropoulos A, Maire P
Development 185975
2019
2018
2017
2016
-
Antisense pre-treatment increases gene therapy efficacy in dystrophic muscles
Peccate C, Mollard A, Le Hir M, Julien L, McGlorey G, Jarmin S, Le Heron A, Dickson G, Benkhelifa-Ziyyat S, Pietri-Rouxel F, Wood M J, Voit T, Lorain S
Hum Mol Genet 25(16): 3555-3563 -
Characterization of a Dmd (EGFP) reporter mouse as a tool to investigate dystrophin expression
Petkova M V, Morales-Gonzales S, Relizani K, Gill E, Seifert F, Radke J, Stenzel W, Garcia L, Amthor H, Schuelke M
Skelet Muscle 6(25) -
Mitochondrial impairment induced by postnatal ActRIIB blockade does not alter function and energy status in exercising mouse glycolytic muscle in vivo
Béchir N, Pecchi E, Relizani K, Vilmen C, Le Fur Y, Bernard M, Amthor Helge, Bendahan D, Giannesini B
Am J Physiol - Endocrinol Metabol 310(7): E539 -
Reorganization of Respiratory Descending Pathways following Cervical Spinal Partial Section Investigated by Transcranial Magnetic Stimulation in the Rat
Vinit S, Keomani E, Deramaudt T B, Bonay M, Petitjean M
PLOS O 11(2): e0148180 -
Therapeutic Potential of Tricyclo-DNA antisense oligonucleotides
Goyenvalle A, Leumann C, Garcia L
J Neuromuscul Dis 3(2): 157-167
2015
-
Caspase-independent apoptosis in infected macrophages triggered by sulforaphane via Nrf2/p38 signaling pathways
Bonay M, Roux A L, Floquet J, Retory Y, Herrmann J L, Lofaso F, Deramaudt T B
Cell Death Dis 1: 15022 -
Dysferlin rescue by spliceosome-mediated pre-mRNA trans-splicing targeting introns harbouring weakly defined 3' splice sites
Philippi S, Lorain S, Beley C, Peccate C, Precigout G, Spuler S, Garcia L
Hum Mol Genet 24(14): 4049-4060 -
Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers
Goyenvalle A, Griffith G, Babbs A, Andaloussi S E, Ezzat K, Avril A, Dugovic B, Chaussenot R, Ferry A, Volt T, Amthor H, Buhr C, Schurch S, Wood M J A, Davies K E, Vaillend C, Leumann C, Garcia L
Nat Med 21(3): 270-275 -
In vivo dynamics of skeletal muscle Dystrophin in zebrafish embryos revealed by improved FRAP analysis.
Bajanca F, Gonzalez-Perez V, Gillespie S J, Beley C, Garcia L, Theveneau E, Sear R P, Hughes S M
Elife 4: e06541 -
Nrf2: new insight in cell apoptosis
Bonay M, Deramaudt T B
Cell Death Dis 6: e1897 -
Un nouvel outil pour le traitement de la myopathie de Duchenne : les tricyclo-ADN
Goyenvalle A, Griffith G, Avril A, Amthor H, Garcia L
Med Sci (Paris) 31(3): 253-256
2014
-
Interdisciplinary approaches of transcranial magnetic stimulation applied to a respiratory neuronal circuitry model
Vinit S, Keomani E, Deramaudt T B, Spruance V M, Bezdudnaya T, Lane M A, Bonay M, Petitjean M
PLOS O 9(11): e113251 -
Myostatin is a key mediator between energy metabolism and endurance capacity of skeletal muscle
Mouisel E, Relizani K, Mille-Hamard L, Denis R, Hourde C, Agbulut O, Patel K, Arandel L, Morales-Gonzalez S, Vignaud A, Garcia L, Ferry A, Luquet S, Billat V, Ventura-Clapier R, Schuelke M, Amthor H
Am J Physiol Regul Integr Comp Physiol 307(4): R444-R454